Cervical Cancer, Childhood
Introduction:
Approximately 1.2 million new cases of invasive cancers are diagnosed annually in the United States. More than 12,000 of these cases involve children.1 The heterogeneity of pediatric cancer is substantial, and even the most common pediatric cancer (ie, acute lymphoblastic leukemia [ALL]) is characterized by biological and clinical diversity. As a result of this heterogeneity and low incidence, the ability of epidemiologists to ascribe causes to specific childhood cancers is extremely limited.
Epidemiological studies have noted the effects of cancer genetics, defined family pedigrees and penetrance, and identified subsets of certain cancers and their implications for treatment and prognosis. In addition, the study of obscure genetic diseases that increase the risk of malignancy in childhood has led to an understanding of important cancer genes, which has wide applicability to oncology in both children and adults.
Tools of Study Measures
The understanding of the epidemiology of any medical problem demands the use of basic terminology from the language of statistics. Important terms are defined as follows:
Ratio - Relationship between 2 quantities (ie, x/y)
Proportion - Ratio in which the denominator also includes the numerator (ie, x/[y + x])
Rate - Proportion occurring per time period (ie, x/[x + y]/time)
Incidence - Proportion of new cases within a population over time (ie, x/[y + x]/time)
Prevalence - Number of existing cases in a population at a set time
Crude rate - Measure of actual events in a population
Standardized rate - Crude rate adjusted for a factor in the population (eg, age, sex, economic status)
Standardized mortality-incidence ratios - Observed rates adjusted by comparison with the expected rate derived from a large population Relative risk - Incidence in a population with a specific characteristic compared to that in a population without the characteristic
Study Designs
Study designs useful to epidemiology include the following:
- Descriptive design - Used to define the characteristics of a particular disease entity
- Ecological design - Used to compare large populations (eg, populations of nations)
- Prospective design - Used to identify 2 similar populations to be treated in different ways in the future for subsequent analysis
Retrospective design - Used to identify and analyze 2 similar populations that were treated in clinical trials; administered by individual institutions, by consortia, and by national and international clinical trial groups, often with oversight by the National Cancer Institute (NCI) of the National Institutes of Health (NIH) and the US Food and Drug Administration (FDA); drug development monitored by The Cancer Therapy Evaluation Program (CTEP) of the NIH
Clinical Drug Trials
New cancer drugs were historically adapted for pediatric use after they were first used in adult patients. New drug development has recently incorporated pediatric trials performed after adult trials, which follow research and development in private industry and academia.
A typical protocol for a clinical trial includes the following information: objectives of the trial, background, patient eligibility criteria, study design, treatment plan, drug information, treatment evaluation criteria, data collection methods, plan for statistical analysis, consent form to be signed by the patient (or parent or guardian) and the investigator, assent by a minor patient, supporting references, and relevant appendices.
Diagnosis and Treatment:
Phase 1 trialsPhase I trials are specifically designed to assess toxicity and thus appropriate dosing. Pediatric patients are treated in cohorts of 3 starting at a dose that is either 75% the adult dose or 10% the lethal dose in mouse studies. The dose is increased in predetermined steps for each new cohort of patients. Toxicity is assessed in several body systems, and a level of dose-limiting toxicity (DLT) is defined. If DLT occurs in at least 1 of the 3 patients, the protocol drops back to the previous dose level, unless the DLT involves only hematologic toxicity in a patient with a hematologic malignancy. Alternative means of identifying dosage can come from studies of toxicity in animals, using a modified Fibonacci scheme. Another method uses continual reassessment in order to escalate to potentially efficacious dosage, risking the encounter of toxicity. Reliance on adult experience is helpful, but obviously not a given, in pediatric-only disease.
Phase 2 trialsPhase 2 trials are typically designed to directly assess the efficacy of a drug in different tumor types. A dose presumed to be safe from the results of phase 1 trials is used. An objective measure of response, such as percentage decrease in tumor size on radiographic imaging or remission assessment within a given period of time during induction, is used to evaluate efficacy. Phase 2 trials typically involve a 2-stage process to establish a firm likelihood and then to measure small differences using statistically significant sample sizes
Phase 3 trialsPhase 3 studies are intended to test the efficacy of novel ways of using drugs (eg, combination chemotherapy, neoadjuvant therapy, timing variations, dose intensification) compared with standard therapy or the natural history of the disease. In addition, this may include a novel drug in combination with accepted therapy as a therapeutic window to assess efficacy in a combined phase 2/3 approach. The design must allow investigators to measure and account for potential false-positive and false-negative data. The potential for error can be calculated and used to decide on the number of patients who need to be enrolled to ensure a certain level of confidence in the results.
A type I error occurs when the P value suggests that a proposed treatment is better than the standard treatment when it is not. The P value is the probability of obtaining the observed data (or data that are more extreme) if the null hypotheses were exactly true. Phase 3 trial design can be sequential to allow data to be continuously evaluated to find effective treatments as quickly as possible. However, type I errors can be magnified in this type of trial. This phenomenon can be ameliorated by requiring increased significance for the study. Factorial designs can examine several factors using a randomization method. Equivalence trials can be designed to determine if a treatment strategy of reduced duration and dosage is as effective as standard therapy.
A key element in phase 3 trials is the inclusion of at least one question that is randomized among alternative possibilities. The randomization mechanism ensures that patients are allocated to respective arms without bias. A method of allocating patients based on random numbers removes predictability from the assignment. Stratification is also desirable to group patients with identifiable prognostic characteristics.
Phase 4 trialsIn phase 4 trials, investigators apply positive findings from research centers to generic use in the community. Phase 4 studies can include large-scale population analyses for marketing and drug promotion by a company or for surveillance as mandated by the FDA. Phase 4 trials can also be conducted for safety and efficacy analysis of already approved drugs. These trials can include the use of controlled randomized studies to study different preparations of already approved agents.
Cancer Incidence
Incidence and mortality rates of childhood cancers differ worldwide. The differences depend on how extensively data are reported. Incidences vary from as high as 155 per million persons in Nigeria to 40 per million persons in the Indian population of Fiji. Rates for the United States are likely to be more accurate than these because 94% of all patients with cancer are reportedly seen at one of the institutions of the Children's Oncology Group (COG).
In the United States, the incidence of childhood cancer overall is approximately 125 per million persons, with slightly increased rates in males and white children. Leukemias account for approximately 25% of all childhood cancers, followed by tumors of the CNS (17%), neuroblastoma (7%), non-Hodgkin lymphoma (NHL) (6%), Wilms tumor (6%), Hodgkin disease (5%), rhabdomyosarcoma (3%), retinoblastoma (3%), osteosarcoma (3%), and Ewing sarcoma (2%). Numerous rare tumor types account for the remainder. These numbers reflect patients in the 0-15 range.
The decreased mortality rate of pediatric cancers has been one of the major success stories of medicine in the last 30 years. Improvements in the survival rates of leukemias, Hodgkin disease, and sarcomas have been notable successes. Most of these improvements can be traced to the use of aggressive multimodal therapy and the judicious use of blood products, use of cytokines, and improved supportive care to prevent and treat infections.
The success of the treatment of pediatric cancer engenders the new challenge of caring for the growing number of cancer survivors. The risk of a second cancer appearing within 20 years after an initial diagnosis of cancer is approximately 8%. The existence of this group also suggests that risk factors (eg, treatment, heredity, other environmental factors) might be identifiable. For instance, the risk of acute myelogenous leukemia (AML) with the 9;11 translocation is approximately 3-6% within 5 years of therapy that includes high-dose etoposide or alkylating agent therapy, depending on dosage and tumor type.
Tumors Leukemias
Leukemias are the most common type of childhood cancer, accounting for 25% of new diagnoses. The greatest advances in treatment and outcomes have occurred in leukemias, in no small part because of the ability to treat relatively large numbers of patients with uniform treatment protocols.
Acute lymphoblastic leukemia
Nearly 80% of childhood leukemias are acute lymphoblastic leukemia (ALL). The advent of modern molecular techniques has resulted in the further dissection of ALL into several subtypes with therapeutic implications. For example, the recently described TEL-AML1 translocation is present in approximately 20% of pediatric cases of ALL. The TEL-AML1 translocation is now considered to be a favorable prognostic indicator for the outcome of ALL, whereas the presence of Philadelphia chromosome, a 9;22 translocation involving the bcr and abl oncogenes, is a poor prognostic indicator.
Although ALL incidence peaks in the first 5 years of life, it represents an even greater number of leukemia cases in older children because the number of acute myelogenous leukemia (AML) cases further decline. Overall, ALL is more common in whites, boys, and in the developed world.
Predisposing genetic conditions are associated with ALL, including Down syndrome, Bloom syndrome, Wiskott-Aldrich syndrome, ataxia telangiectasia, and other immunodeficiency syndromes. Environmental factors have also been implicated. Radiation exposure has been most tightly linked to ALL in children. Although the effect of radiation has been difficult to quantify, the risk of in utero exposure has been reported to be significant.
Acute myelogenous leukemia
Approximately 18% of childhood leukemia cases involve AML. This ratio of ALL-to-AML remains constant throughout childhood, except for a predilection for AML in the neonatal period. AML comprises a heterogeneous array of subtypes. Molecular diagnostic methods have advanced the ability to subtype myeloid leukemias; the analysis of translocations is helping to define and confirm the histologic designations.
For example, the t(8;21) translocation is found in 15% of patients with AML. Of interest, this translocation is a favorable predictor of long-term survival. The acute promyelocytic subtype, which is associated with a 15;17 translocation, is similarly correlated with a favorable outcome by virtue of its response to therapy with all–trans-retinoic acid. In contrast, the chromosome 9;11 translocation associated with the monocytic subtypes indicates a poor prognosis. Abnormalities of chromosome 11 at the malignant lymphoma, lymphoblastic (MLL) locus are often observed in individuals with secondary AML after treatment with etoposide. Abnormalities in the oncogene, FLT-3, such as internal tandem duplications and loss of whole or parts of chromosomes 7 and 5, are associated with poorer outcomes.
Exposure to ionizing radiation in Japan resulted in increased AML risks. Organic solvent exposure has also been associated with AML. Exposure to chemotherapeutic agents such as Topo II drugs and alkylators also predispose to secondary AML. A long list of genetic diseases are also associated with AML.
Chronic leukemias
Chronic leukemias account for less than 5% of all pediatric leukemias. Chronic myelogenous leukemia (CML) is the most common type and corresponds to the adult type of CML marked by the Philadelphia chromosome. This adult type of CML appears in children younger than 4 years and is linked to radiation exposure in many individuals with CML. Juvenile myelomonocytic leukemia (JMML), a chronic leukemia that lacks the bcr-abl fusion, is a disease found in younger children, with most cases diagnosed in children younger than 2 years. Other rare forms of chronic childhood leukemia include chronic myelomonocytic, monocytic, and lymphocytic leukemias.
Brain Tumors
Tumors of the CNS constitute the other major type of childhood cancer. Roughly 20% of childhood cancers involve brain tumors. Patients with CNS tumors remain an underreported segment of the pediatric population with cancer because only one half are referred to specialty centers. Morbidity is clearly the greatest problem in patients with brain tumors because many of these tumors are in locations that are difficult to treat. Most pediatric brain tumors occur in the first decade of life. Unlike adult brain tumors, most true childhood brain tumors occur in the posterior fossa.
Brain tumors are heterogeneous, which makes their overall classification difficult. The most common brain tumor in children is medulloblastoma, which accounts for 10-20% of childhood brain tumors and 40% of tumors in the posterior fossa. Most brain tumors, chiefly medulloblastomas and glial tumors, involve the posterior fossa after the first 2 years of life. Most CNS tumors are glial tumors, which are classified by their location as supratentorial, cerebellar, or brainstem. Supratentorial astrocytomas comprise 30-40% of cases, with cerebellar astrocytomas and brainstem gliomas (15% each) comprising the remainder of the glial tumors. Unique variants in each of these groups have strong prognostic significance. For example, patients with exophytic gliomas do extremely well, whereas individuals with diffuse infiltrative tumors do poorly.
Various genetic syndromes predispose probands to brain tumors, including neurofibromatosis, Li-Fraumeni syndrome, and tuberous sclerosis. Environmental exposure and immunosuppression are also associated with increased risk, including radiation (gliomas) and HIV (lymphoma).
Hodgkin Disease
Rates of Hodgkin disease, which accounts for 5% of childhood cancers, peak in children younger than 14 years, in young adults, and in adults older than 55 years. Most statistical reports comment on childhood cancers in individuals aged 14 years or younger. Like non-Hodgkin lymphoma (NHL), Hodgkin disease is reported to be associated with immunodeficiency and infection with the Epstein-Barr virus (EBV), as well as cytomegalovirus.
Classification of Hodgkin disease includes specific subtypes, including nodular sclerosing, lymphocyte predominant, mixed cellularity, and lymphocyte depleted. Nodular sclerosing appears to be the most common subtype, and lymphocyte depleted seems to be associated with severe disease and worsened outcomes. Patients who survive Hodgkin disease remain at high risk for secondary tumors, a phenomenon that may indicate an underlying immunodeficient state. Breast cancer in young patients with a history of Hodgkin disease is mostly associated with irradiation as a treatment modality.
Neuroblastoma
Neuroblastoma is the most common non-CNS solid tumor. Both long-term survival and short-term treatment remain challenges in the care of patients with neuroblastoma. Of interest, the patient's age at presentation has prognostic implications. The type that emerges in infancy greatly improves the likelihood of long-term survival and is marked by a lack of N-myc amplification; by hyperdiploidy; by low-stage, limited distant sites in stage I or II disease (marrow, liver, or skin involvement in < 10% of patients); by the absence of 1p chromosomal abnormalities; by a lack of changes on chromosome 17; and by evidence of neuronal differentiation. However, the form that emerges in children aged 1-10 years has a much worse prognosis. Association with genetic alterations have been characterized, including germline mutations in the ALK gene and chromosome 1p deletions.
Non-Hodgkin lymphoma
Lymphomas make up a large, if heterogeneous, category of childhood cancers. Chief among these cancers are the NHLs, which are responsible for 6% of all pediatric cancers. NHL is a disease of young children and is more prevalent than Hodgkin lymphoma in the first decade of life; it has an overall predilection for boys, probably because of a subset of T-cell lymphoma. A major factor in NHL is its association with immunodeficiency secondary to underlying genetic diseases, viral infection, or drugs. Burkitt lymphoma, roughly 40% of all NHL, is associated with EBV infection and endemic on the African continent. Burkitt lymphoma accounts for roughly one half of all incidents of NHL, a number which translates to an incidence of approximately 2-3% among childhood cancers. In its endemic form, the incidence of Burkitt lymphoma can increase as much as 50-fold. Endemic Burkitt lymphoma is associated with EBV and appears to occur in equatorial Africa. Additional environmental factors appear to be at work in the pathogenesis of Burkitt lymphoma because the endemic form differs from even the sporadic form, which can also be found along with EBV in North America as the breakpoints of the 8:14 translocation differ.
Lymphoblastic lymphoma and large cell lymphoma
Comprising 30% of NHL, most lymphoblastic lymphomas resemble T-cell ALL in epidemiological incidence and male predominance, with a constant incidence amongst age groups. No virus or chromosomal abnormality has been associated with lymphoblastic lymphomas. The large cell lymphomas comprise 30% of NHL cases. Anaplastic (30%), diffuse, and mediastinal lymphomas are observedThis variant is associated with EBV in the setting of HIV.
Renal Tumors
Wilms tumor is the most common renal tumor overall, comprising approximately 5-6% of childhood cancers; however, in infancy, related tumors such as mesonephric nephroma are more common.
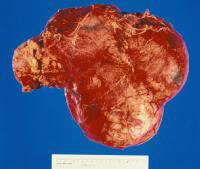 Gross nephrectomy specimen shows a Wilms tumor pushing the normal renal parenchyma to the side. As in neuroblastoma, the patient's age affects the prognosis, in that patients who present in infancy have the best outcomes. Wilms tumor is strongly associated with a host of genetic syndromes, including Beckwith-Wiedemann syndrome; Denys-Drash syndrome; and Wilms tumor, aniridia, genitourinary abnormalities, and mental retardation (WAGR) syndrome. Studies of chromosome 11 have led to the description of the products of the WT1 and WT2 genes, which are associated with WAGR syndrome and Beckwith-Wiedemann syndrome, respectively. Prognostic factors associated with long-term survival include low-stage disease, favorable histology, and young age.
Gross nephrectomy specimen shows a Wilms tumor pushing the normal renal parenchyma to the side. As in neuroblastoma, the patient's age affects the prognosis, in that patients who present in infancy have the best outcomes. Wilms tumor is strongly associated with a host of genetic syndromes, including Beckwith-Wiedemann syndrome; Denys-Drash syndrome; and Wilms tumor, aniridia, genitourinary abnormalities, and mental retardation (WAGR) syndrome. Studies of chromosome 11 have led to the description of the products of the WT1 and WT2 genes, which are associated with WAGR syndrome and Beckwith-Wiedemann syndrome, respectively. Prognostic factors associated with long-term survival include low-stage disease, favorable histology, and young age.
Retinoblastoma
With an overall incidence of around 2%, retinoblastoma is a relatively rare but classic solid tumor. Its study led to the development of the 2-hit hypothesis of carcinogenesis. Studies of family trees and analysis of known mutations have demonstrated an incidence of unilateral plus sporadic (60%), unilateral plus inherited (15%), and bilateral plus inherited (25%). Hereditary retinoblastoma occurs early, often at birth and 80% before age 2 years and is most likely to be bilateral, implying that a second mutation in the RB gene with the first hit having been inherited in the germline.
Incidents of sporadic retinoblastomas are simply most likely to be unilateral by virtue of the lowered likelihood of 2 hits occurring in a normal somatic cell. Inherited retinoblastoma cases illustrate the importance of the Rb protein in suppressing tumorigenesis in that patients with inherited retinoblastoma remain at risk for other tumors, chiefly osteosarcoma.
Rhabdomyosarcoma
Rhabdomyosarcoma, which comprises roughly 3% of childhood cancers, is another solid tumor with an incidence that peaks in children younger than 6 years and again in early adolescence. This incidence is roughly correlated with the type of tumor. Head and neck tumors are generally diagnosed in young patients (two thirds of cases), and the histology is usually embryonal. Older patients (one third of cases) are most likely to have tumors in the extremities with alveolar histology. In general, patients with embryonal tumors and individuals with hyperdiploidy have improved outcomes; however, these data remain somewhat controversial. Associations with Li-Fraumeni syndrome, Beckwith-Wiedemann, and neurofibromatosis have all been reported.
Osteosarcoma
Osteosarcoma is a bone tumor associated with the rapid bone growth characteristic of the adolescent growth spurt. Although more common overall, it is less common than Ewing sarcoma in the first decade of life. Osteosarcoma is most common in patients who are taller than their peers and is diagnosed at an early age in more girls than boys. Tumors are localized to the metaphyseal part of long bones, with most common sites including distal femur (30%), proximal tibia (15%), and proximal humerus (10%). Radiation and alkylating agents have been implicated in the etiology of osteosarcoma, along with retinoblastoma and Li-Fraumeni syndrome.
Ewing sarcoma
Ewing sarcoma represents a group of tumors that includes peripheral primitive neuroectodermal tumors and primary bony tumors. The diagnostic standard involves detection of either the chromosome 11;22 or the 21;22 translocation, at least one of which is found in as many as 95% of individuals with Ewing sarcoma. An interesting feature of Ewing sarcoma is its extreme rarity among blacks and significant occurrence in whites. Although the greatest incidence is observed in the second decade of life, Ewing sarcoma occurs more throughout the age spectrum than does osteosarcoma. Ewing sarcoma is not associated with rapid bone growth and may be found anywhere along the bone or adjacent soft tissue or may even occur as an isolated soft-tissue mass. The most common sites of Ewing sarcomas are the pelvis (26%), femur (20%), tibia (10%), and chest wall (16%).
Cancer Predisposition Factors
Relatively few causative factors have been identified for childhood cancer. The increased numbers of adults with cancer have enabled the ascertainment of causative factors, such as alcohol and smoking, whereas the small numbers of children with cancer have made environmental factors difficult to evaluate. However, analysis for inherited factors is increasingly fruitful, given the explosion in availability of molecular biologic technology and resources engendered by the Human Genome Project.
Inherited Predisposition
At its most basic level, cancer is a genetic disease. Production of genetic instability that confers some kind of mutator phenotype is most likely the chief characteristic of any inherited predisposition for cancer. These instabilities take one of several forms: (1) mutations in key genes that are directly involved in tumoral development (eg, WT1, WT2), (2) mutations in genes that generate mutations and gross chromosomal deletions at key loci (eg, in Fanconi anemia and mismatch repair), (3) mutations in genes directly involved in DNA repair of specific lesions (eg, xeroderma pigmentosum), and (4) complex chromosomal syndromes that increase the person's susceptibility to develop cancer.
Down syndrome
Children with Down syndrome have a 1% risk of developing leukemia before age 10. The ratio of types is different in these children than in children overall in that 60% of children with Down syndrome develop acute lymphoblastic leukemia (ALL), and 40% develop acute myelogenous leukemia (AML). In general, the prognosis in some reported series is no better or worse in children with Down syndrome and ALL than in children without Down syndrome and ALL. In contrast, outcomes tend to be better in children with Down syndrome and AML than in children without Down syndrome and AML. Interestingly, AML in Down syndrome is skewed toward the megakaryoblastic form. Roughly 10% of patients with Down syndrome may have associated transient myeloproliferative disease (TMD) of infancy, which not only resembles congenital leukemia but also confers a 20-30% risk of subsequent AML.
Turner syndrome
Retention of the Y chromosome in female individuals with Turner syndrome mosaicism or androgen insensitivity syndrome increases their lifetime risk of gonadoblastoma. This risk is as high as 25% by adulthood.
Wilms tumor
Association of gross deletions at the 11p13 locus with Wilms tumor led to isolation of the WT1 gene. Clinical abnormalities associated with WT1 mutations include aniridia, genital abnormalities, and mental retardation. As many as 40% of individuals with Wilms tumor have some familial component.
Syndromes associated with increased growth have also been associated with Wilms tumor. Examples include the Beckwith-Wiedemann syndrome and hemihypertrophy. Beckwith-Wiedemann syndrome is linked to chromosomal band 11p15, where a putative WT2 gene resides. Insulin growth factor 2 and p57kip2 are the leading candidates for the WT2 tumor suppressor gene.
Mendelian Inheritance of Genetic Cancer PredispositionAutosomal dominant disorders
In his study of retinoblastoma, Knudson first described the 2-hit hypothesis of carcinogenesis. This hypothesis describes the process whereby, given the genetic transmission of these disorders through the germline, the loss of a second allele of the same gene in a predisposed patient leads to the onset of cancer at an early age. Germline defects in one allele may predispose the person to or may promote the loss of the other corresponding allele. These disorders are more likely than other cancers to be associated with bilateral and multiple tumors. Concomitant with this risk is the person's risk of developing several tumors at various times during his or her lifetime, depending on the tissue at risk.
With regard to retinoblastoma, the deleted RB gene not only increases the risk of the patient born with the mutation but also entails unknown risk for 2 other groups: patients with newly diagnosed sporadic cases and familial carriers who do not develop retinoblastoma as children. Mutation in the RB gene also confers a lifetime risk of osteosarcoma and melanoma.
The p53 gene represents the gene most commonly mutated in human cancers and is the dysfunctional gene responsible for the rare familial Li-Fraumeni cancer syndrome. Numerous cancers cluster in Li-Fraumeni cancer syndrome, including sarcomas, breast cancer, leukemia, brain tumors, and adrenocortical carcinoma. The study of Li-Fraumeni syndrome has improved our understanding of cancer in general because p53 appears to be a convergence point for many cancers in the long, multistep process of carcinogenesis.
The development of several colonic polyps is associated with the early development of familial colon cancer and hepatoblastoma. The APC gene was found by positional cloning. This gene affects cellular signaling pathways by means of the beta-catenin pathway.
Hereditary nonpolyposis colon cancer (HNPCC) was first defined as a genomic instability disorder in which the underlying genetic defect promotes the loss of the other allele, giving rise to the tumor. The HNPCC group involves at least the mismatch repair proteins that are implicated in an array of adult cancers. The mismatch repair genes are analyzed at the protein level and as an in vitro test to determine a person's carrier status for HNPCC.
The gene complex for multiple endocrine neoplasia (MEN) is marked by an association of cancers of the thyroid, parathyroid, pancreas, pituitary, and adrenal medulla. MEN type 2 syndrome appears to be due to activating mutations of the ret oncogene rather than to a 2-hit mechanism.
Neurofibromatosis type 1 is one of the most common genetic syndromes and is marked by a propensity to cause brain tumors and peripheral nerve sheath tumors. Mutations in the ras guanosine triphosphatase (GTPase) gene, termed neurofibromin, are sporadic in at least one half of the cases of neurofibromatosis detectable in the general population. The frequency is 1 case per 3000 persons. Patients with neurofibromatosis type 1 are prone to develop optic gliomas, most commonly in early childhood, along with gliomas in other locations. A link to the development of myeloid leukemias is also described; this link is consistent with the connection between ras mutations and myeloid disease. Associations with many other diseases are reported but not proven.
Tuberous sclerosis is a syndrome of seizures, mental retardation, and angiofibromas. Tuberous sclerosis is associated with a range of benign growths. Cardiac rhabdomyomas are a problem of infancy, whereas retinal hamartomas and giant cell astrocytomas develop later in childhood.
Free Consultation
Conditions